Whole Plasmid, ZeroPrep, & RCA Sequencing Services
Lots of reasons!
- Scientific rigor and peace of mind.
- E. coli and other hosts will go to great lengths to avoid expressing your leaky toxic gene, including modifying your plasmid in unexpected ways that are invisible to targeted Sanger sequencing.
- Plasmid inserts are getting longer and more complex. Instead of multiple Sanger runs or synthesizing a sequencing primer or doing primer walking, sequence the whole plasmid.
- Long reads are ideal for resolving repetitive regions that stymie Sanger sequencing.
- Are you sure your plasmid isn't a dimer? Are you sure there aren't multiple plasmids in your strain? Sanger sequencing won't tell you, and we see it all the time.
- It's neither much more expensive nor slower.
We sequence each sample with Oxford Nanopore long reads to very high depth before generating a consensus/assembly using the latest basecalling and polishing software:
- We construct an amplification-free long-read sequencing library using the newest v14 library prep chemistry, including linearization of the circular input DNA in a sequence-independent manner.
- We sequence the library with a primer-free protocol using the most accurate R10.4.1 flow cells (raw data is delivered in .fastq format).
- We generate a high-accuracy circular consensus sequence from the raw reads.
- For standard size plasmids, we will also return a set of feature annotations.
Plasmid and circular samples are sequenced WITHOUT primers or amplification. Please do not ship any primers with your samples or mix primers into your samples.
In the vast majority of cases, we deliver plasmid sequencing results within one business day of receipt of your samples.
This service is intended for a clonal population of molecules. You can send mixtures of molecular species, but since we can't predict the analysis outcome, it's at your own risk.
- If your species are very similar (e.g. differ by only a few nucleotides), the pipeline will most likely create a single .gbk consensus file, with mixed peaks observed in the .ab1 file at SNP/indel locations.
- If your species are sufficiently distinct (e.g. vastly different in size or sequence), the pipeline will generate a single consensus sequence for the molecular species that produces the largest amounts of total sequencing data. (Note that concatemer forms such as dimers, trimers, etc. are not considered different molecular species by the pipeline, so you will only receive the monomer consensus sequence by default).
Ultimately, which species ends up producing a consensus will vary depending on overall sample quality, coverage, and relative abundance/degradation of each species.
Sequencing is considered successful if the pipeline is able to generate any consensus, even if it is not your target. Re-sequencing mixtures won't change the relative proportions of the species (and thus which species generates a consensus), but you can submit multiple aliquots if you need higher overall coverage.
If you'd like to sequence a known mixture (e.g. barcode or variant libraries), please consider submitting instead to our Custom Sequencing Service.
As per Oxford Nanopore's specs for the chemistry and flowcells we currently use for plasmid sequencing, the consensus accuracy is typically >99.99%.
The most common error modes for Oxford Nanopore are deletions in homopolymer stretches (especially if longer than 8 bp), errors at the Dam methylation site GATC, and errors at the middle position of the Dcm methylation site CCTGG or CCAGG. These limitations are expected to improve with future updates to ONT sequencing chemistry and basecalling software.
We do not guarantee any specific level of coverage, as the number of raw reads generated can vary substantially depending on sample quality.
Successful samples sent at the required concentration typically yield in the high dozens to hundreds (or thousands!) of raw sequencing reads.
Average coverage is reported in the SAMPLE_summary.tsv file. Coverage over ~20x indicates a very accurate consensus.
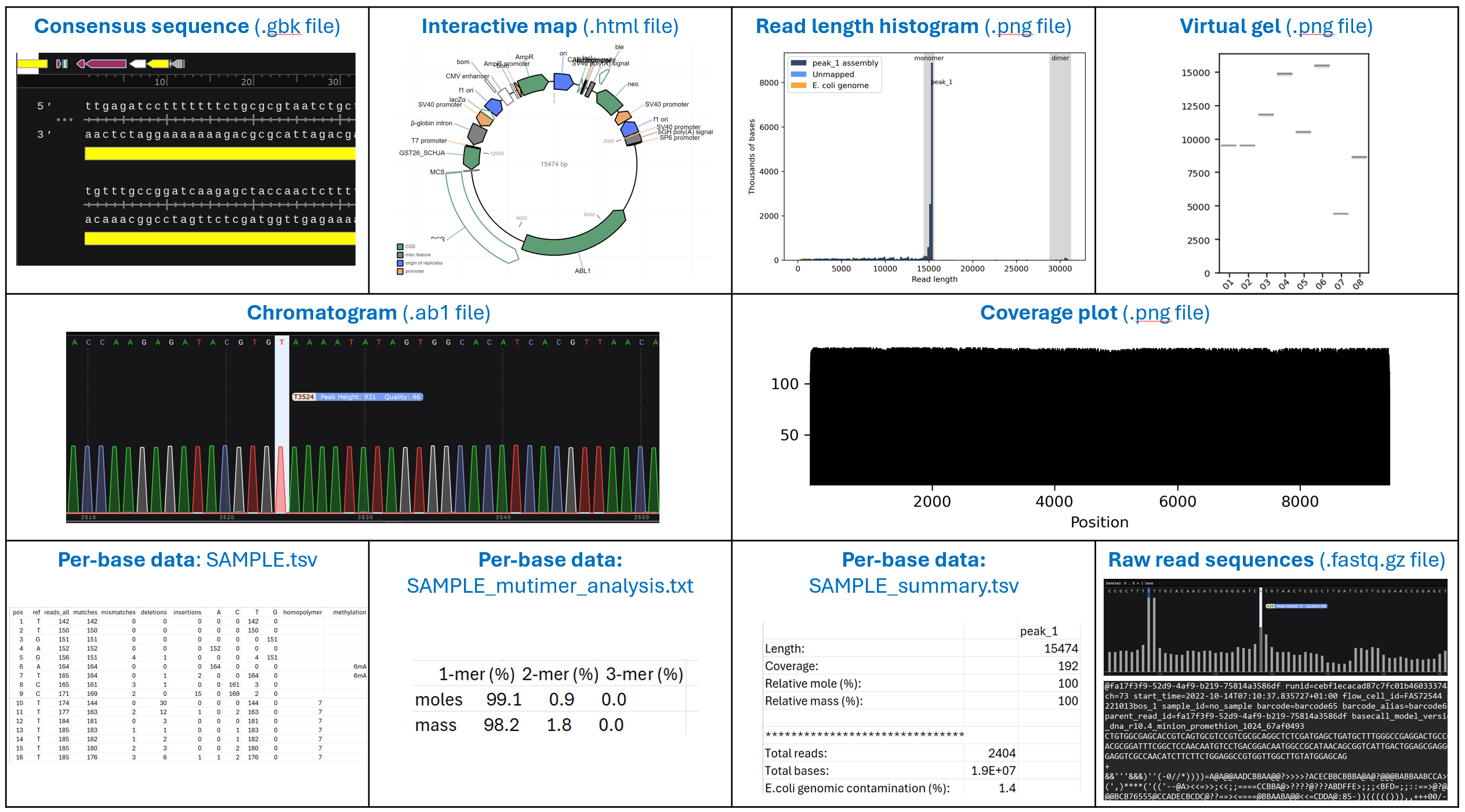
- Consensus sequence (.fasta file): Provides the polished consensus sequence of the plasmid, generated from the raw reads.
- Consensus sequence (.gbk file): Provides the polished consensus sequence of the plasmid, generated from the raw reads. Also includes a plasmid map and feature annotations from the excellent pLannotate tool from the Barrick Lab:
McGuffie,M.J. and Barrick,J.E. (2021) pLannotate: engineered plasmid annotation. Nucleic Acids Research DOI: 10.1093/nar/gkab374 - Plasmid map (.html file): An interactive version of the pLannotate plasmid map.
- Read length histogram (.png file): Displays the read length distribution of the raw reads produced by your sample, thereby providing unique insight into the contents of your samples. See more details about how to interpret your histograms below.
- Virtual gel (.png file): Displays the raw read lengths from all samples in the order in a virtual gel format, resembling what you’d see if you ran the DNA fragments on a gel.
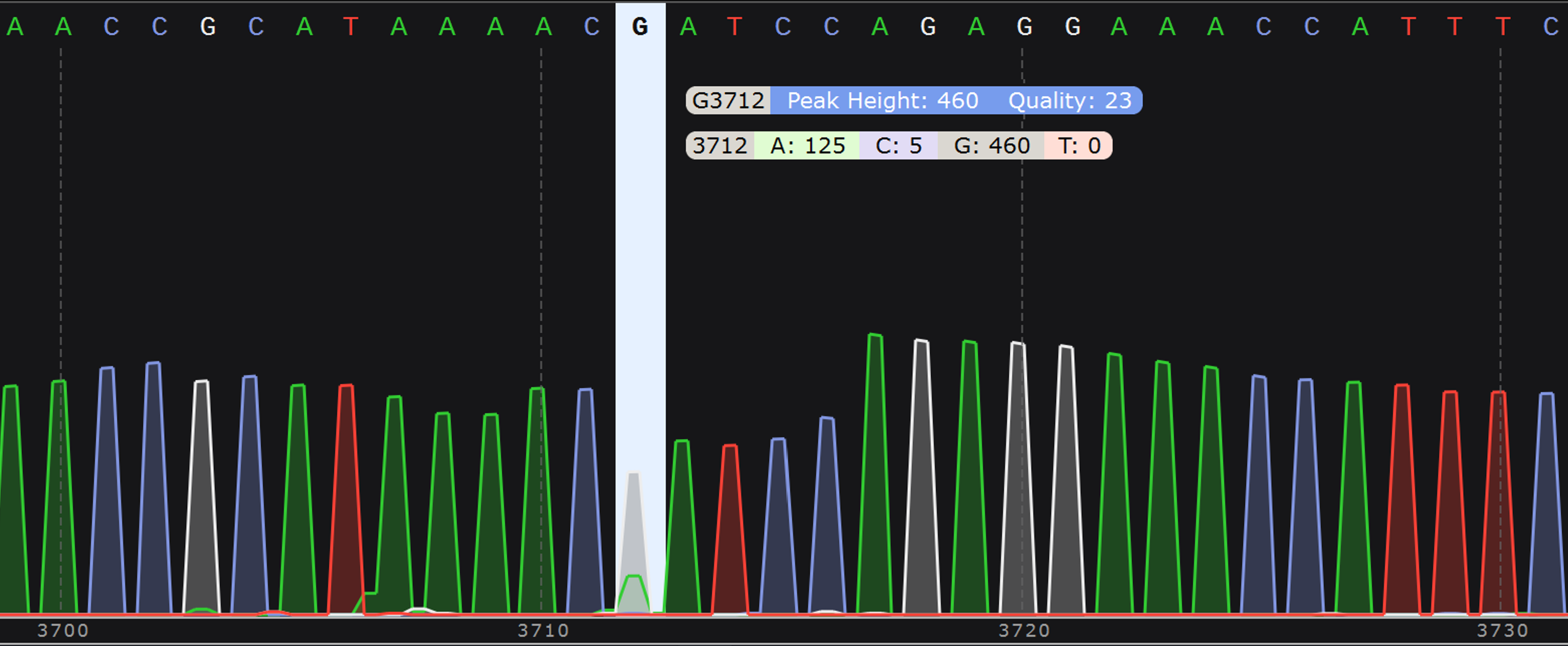
- Chromatogram (.ab1 file): Displays the relative abundance of each nucleotide (A, T, G, C) for all raw reads that align to the consensus at each position of the sequence. See more details about how to interpret your chromatograms below.
- Coverage plot (.png file): Displays the relative sequencing coverage at each position of the consensus sequence. A region with a large gap or a sudden large increase suggests either an assembly issue or a mixture of multiple plasmid species.
- Per-base data (.txt and .tsv files): Includes 3 sub-files for each sample:
- SAMPLE.tsv: Indicates how well the raw reads agree with the consensus sequence at each position. The list includes the consensus basecalls at each position, along with number of total raw reads aligning at that position and the basecall distributions in the raw reads for that position (A, T, G, C, matches, mismatches, insertions, deletions, etc.).
- SAMPLE_multimer_analysis.txt: Indicates the % distribution of the various concatemer forms of the consensus sequence (monomer, dimer, trimer, etc.).
- SAMPLE_summary.tsv: Indicates the length, average coverage, relative composition (by moles and mass), total reads, total bases, and %. E. coli genomic DNA contamination for the consensus sequence.
- Raw read sequences (.fastq.gz file): Provides the sequences of individual raw reads that align to the consensus. Please note that these reads are NOT delivered in the default download, but can be downloaded separately by clicking the "Download Raw FASTQ" button at the top of the "Order Information" page. Note that any raw reads that do not align to the consensus (e.g. host genomic DNA, lower abundance molecular species) are excluded.
FASTQ is a sequence format similar to FASTA, with additional Phred quality score information that can displayed graphically by most modern sequence viewers. Since each basecalled position only has one quality score, certain sequence features, such as insertions or deletions, must be inferred from looking at adjacent bases.
Our ability to deliver these target outputs is directly dependent on the quantity, quality, and purity of the linear/PCR DNA sent to us, so we do not guarantee results. If we are not able to generate a consensus sequence from your sample, our failure policy applies.
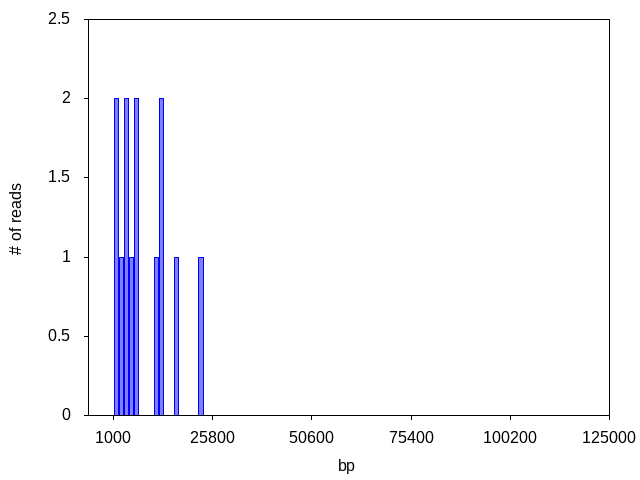
The histogram displays the lengths of the raw reads produced by your sample, with read length (bp) on the x-axis and thousands of bases of data collected (kb) at that length on the y-axis. The histogram is therefore weighted by amount of sequencing data produced by different sizes of molecules; for example, two DNA fragments of different lengths that produce the same number of reads will produce different amounts of total data.
The x-axis is automatically scaled to the maximum read length produced by your sample. Before sequencing your plasmids, we linearize them so that we get mostly full-length sequence reads. As a result, the lengths of the raw sequencing reads reflect the lengths of the molecular species in your sample.
Additionally, the histogram color key indicates what fraction of the raw data maps to the consensus sequence:
| The data from a raw read is colored as... | If... |
|---|---|
| Dark blue (ASSEMBLY read) | Raw read aligns to the consensus/assembly sequence |
| Orange (E. COLI read) | Raw read aligns to the E. coli genome |
| Light blue (UNMAPPED read) | Raw read does not align to any of these categories (Could be sequencing noise, a genome other than E. coli, a lower abundance plasmid species that does not generate a consensus, etc.) |
Ideally, your target plasmid will be the only species in the sample, and we will see one dominant peak in the read length histogram:
(Please note that even a single apparent peak MAY contain multiple plasmids of the same size, or multiple plasmids of different lengths that happen fall into the same histogram bin. Sequences that are very similar are assumed by the analysis pipeline to be variations of a single species and it will attempt to make a single consensus (with potentially low confidence positions reported); if the sequences are very distinct, it will only produce a consensus for the most abundant species.)
If your raw reads contain varying numbers of indels (common for noisy raw reads), this may sometimes cause the read lengths to straddle a bin boundary and artifactually create an appearance of two separate peaks:
(Please note that a peak straddling a bin boundary MAY contain multiple plasmids of the same size, or multiple plasmids of different lengths that happen fall into two adjacent histogram bins. Sequences that are very similar are assumed by the analysis pipeline to be variations of a single species and it will attempt to make a single consensus (with potentially low confidence positions reported); if the sequences are very distinct, it will only produce a consensus for the most abundant species.)
More often than you would expect, though, we see multiple peaks corresponding to multiple plasmids, or a peak of a different size than the customer expected:
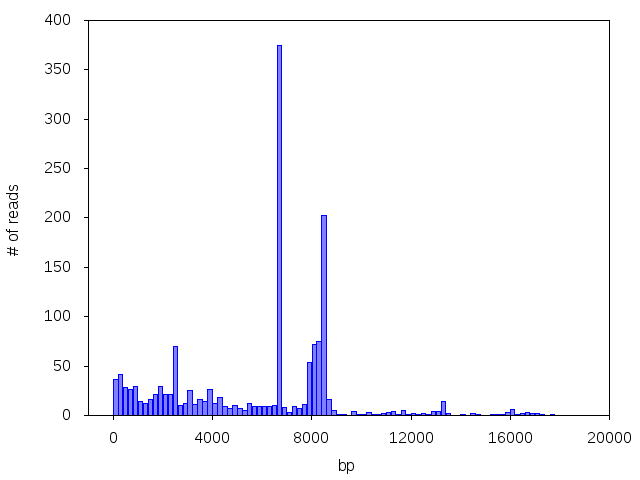
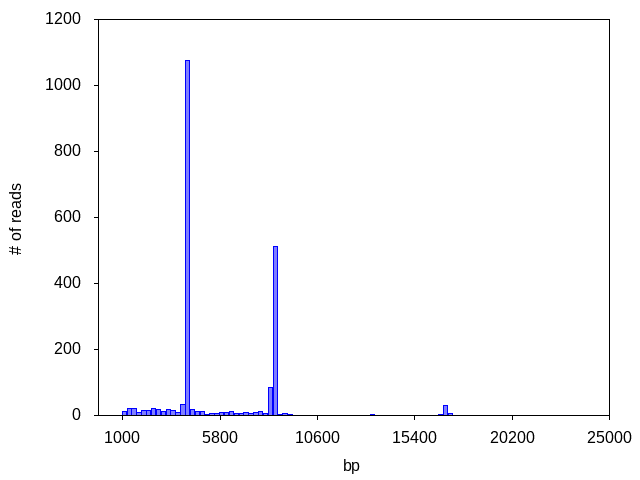
If you sample contains a mixture, we will return only a single consensus for the molecular species that produces the largest amount of total sequencing data. If you’d like us try generating a consensus for an alternate peak instead, you can email us at support@plasmidsaurus.com to inquire.
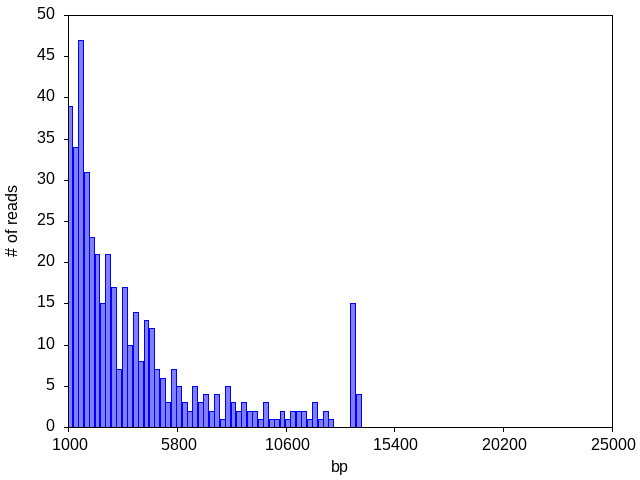
Occasionally we see a sample with a dominant peak in addition to an abundance of degraded DNA (genomic and/or plasmid). In some cases the dominant peak may still produce a consensus, if read coverage and accuracy are sufficient:
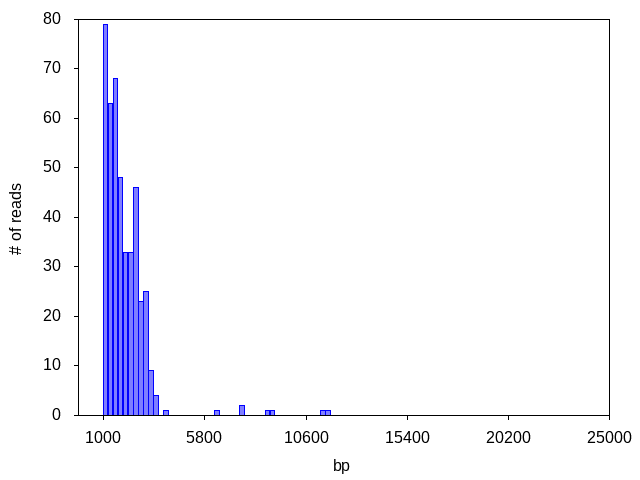
Sometimes we see a decent number of reads for the sample but there is NO dominant peak, indicating an abundance of degraded DNA (genomic and/or plasmid) from a poor plasmid prep, or that the strain contains no plasmids:
Often, the read count is too low to distinguish any peaks or to generate any consensus:
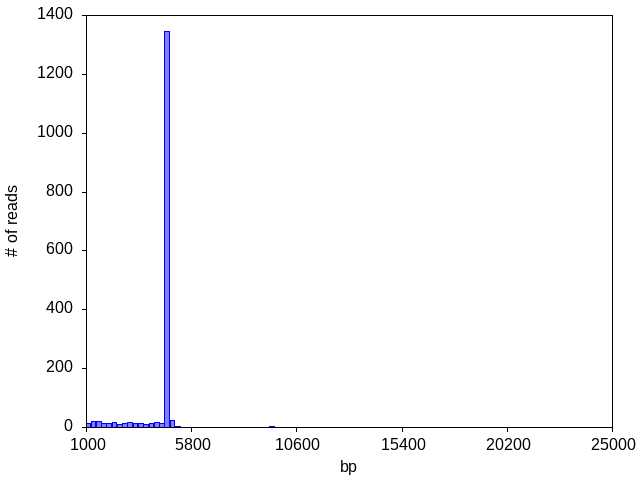
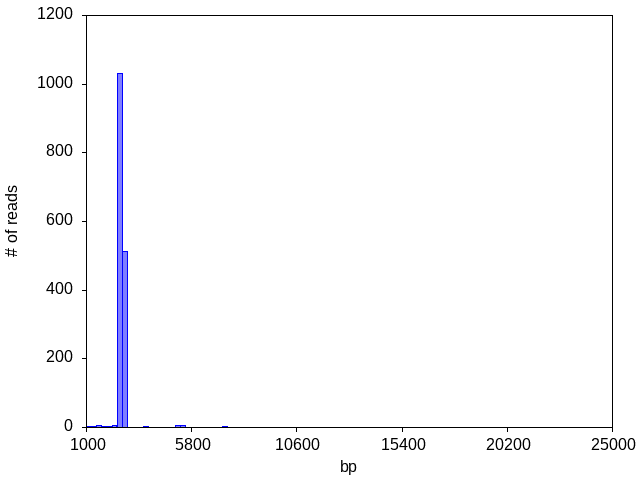
We see concatemers like this all the time -- they are not a sequencing artifact. Sanger sequencing can't detect them and you won't see them on gel of your digested/linearized plasmid, so you're not used to seeing them, but they turn out to be very common. If you run your sample uncut on a gel with a supercoiled ladder, you will see the concatemer band.
They often seem to be formed in vivo during growth in a RecA+ strain (such as NEB Turbo cells), and are more common when plasmids have large repetitive regions or other complex structures. Even plasmid manufacturers like Addgene observe that concatemers occur frequently, and that only the long-read sequencing technologies like the one we use here Plasmidsaurus (that is, Oxford Nanopore Technologies) can detect them!
Please note that concatemer forms such as dimers, trimers, etc. are not considered different molecular species by the pipeline, so you will only receive the monomer consensus sequence by default, even if other concatemer forms produced more sequencing data.
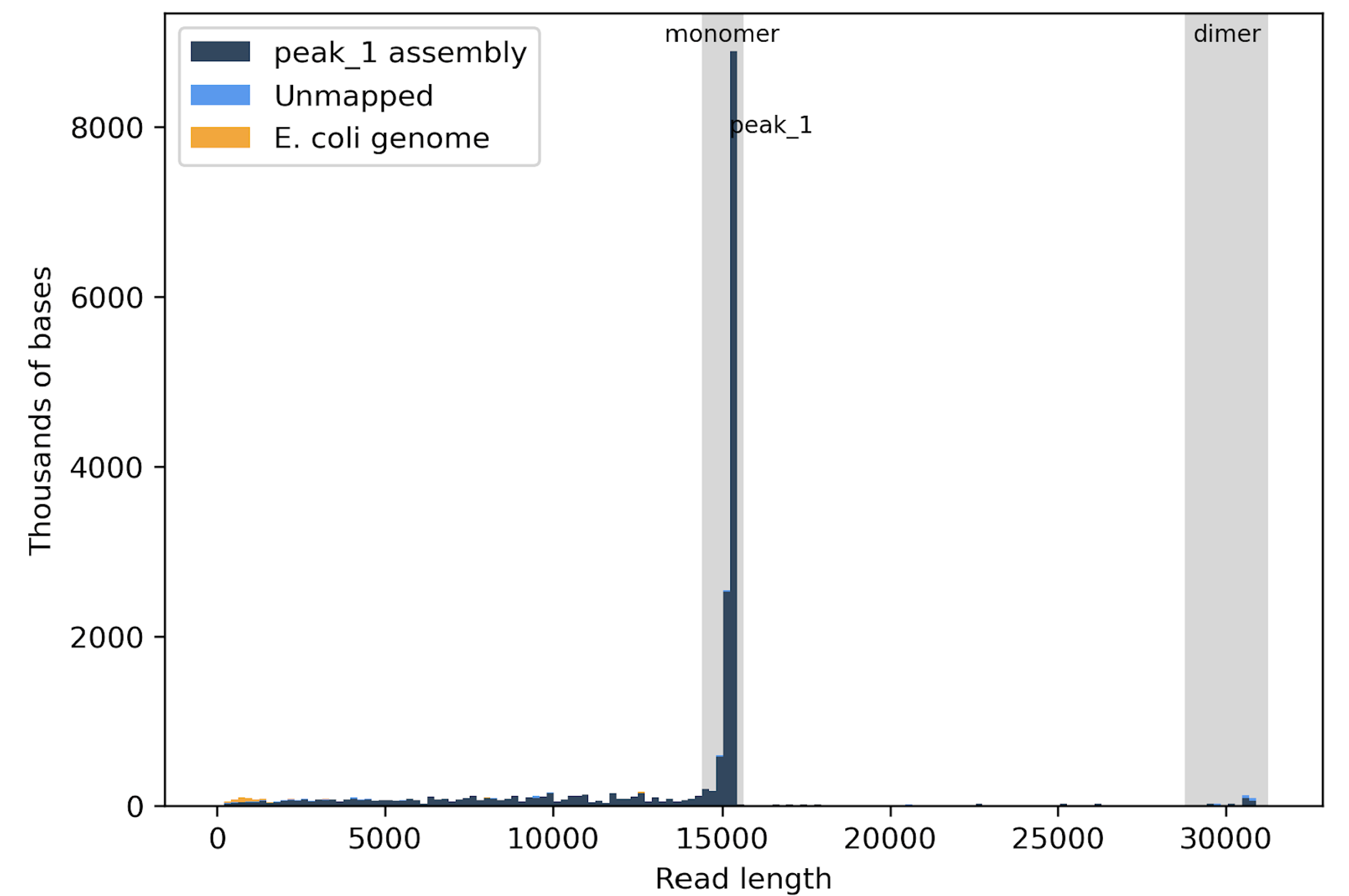
Fig1. This histogram shows that most of the data is produced by the monomer, but these is also a small amount of data from the dimer.
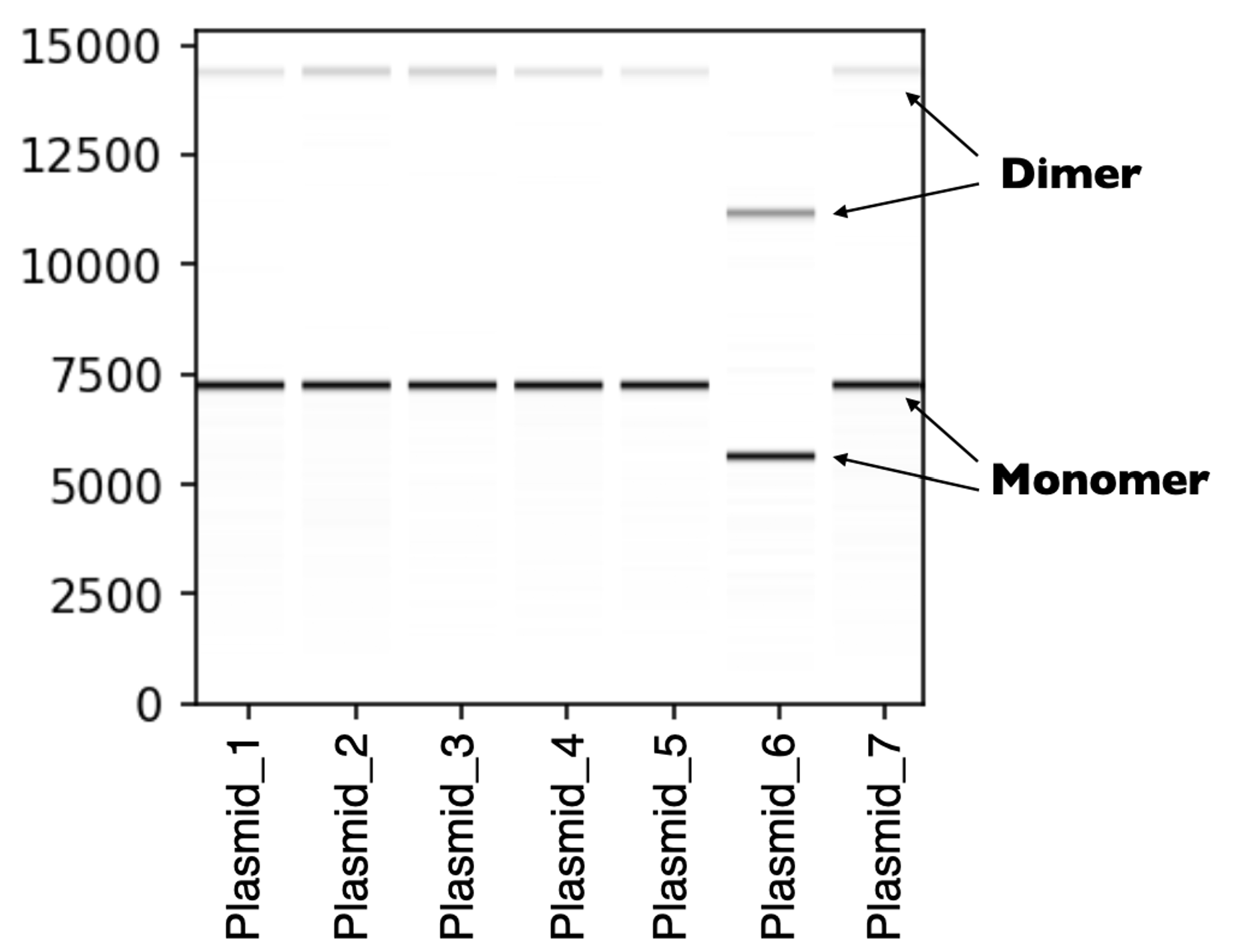
Fig2. Each sample shown in this virtual gel displays two distinct bands, one for the monomer and one for the dimer.
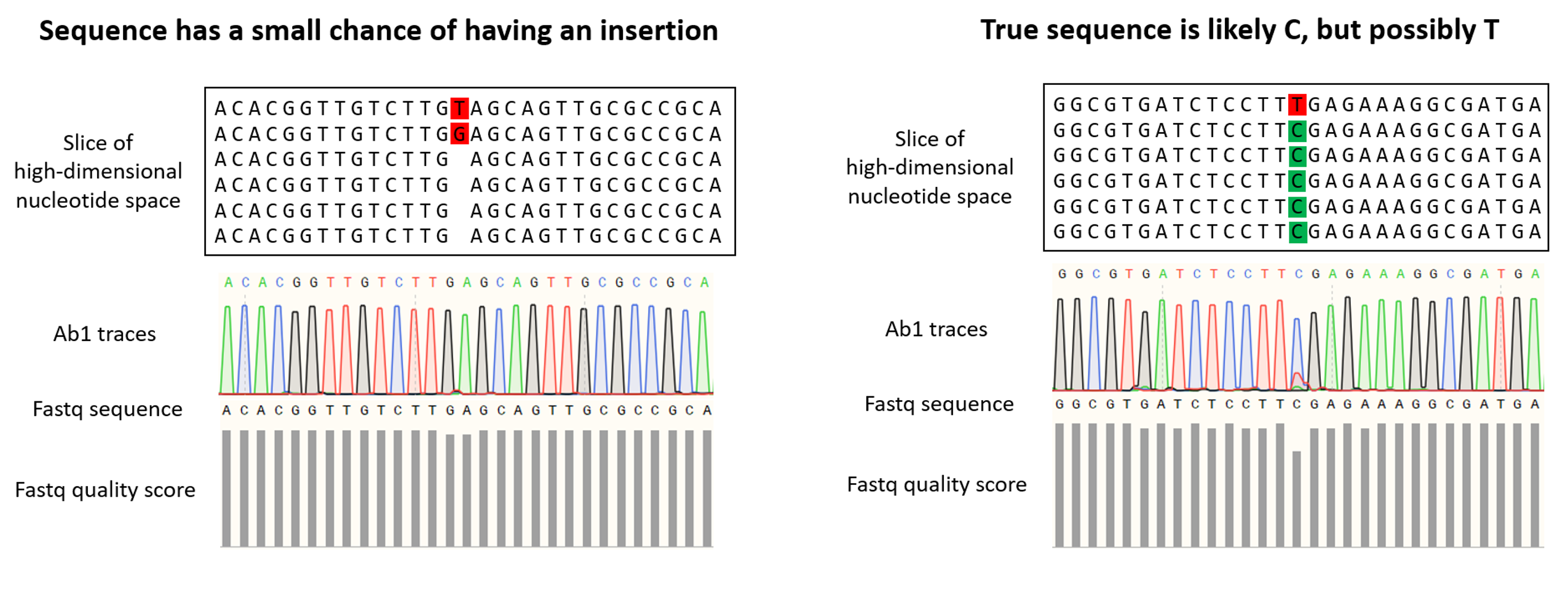
The .ab1 format has widespread use in Sanger sequencing and normally indicates the intensity of fluorescent nucleotides (A, T, G, C) at each position of the consensus. Since fluorescence is not employed in the Oxford Nanopore Sequencing technology that we use here at Plasmidsaurus, we generate this .ab1 file synthetically using the relative abundance of each nucleotide (A, T, G, C) from the raw reads at each position of the consensus sequence. Because this file type was originally used for Sanger sequencing (which is limited to much shorter read lengths than we get with Oxford Nanopore), the file has a maximum size limit and therefore we must often report sequences in multiple pieces.
This file gives a visual representation of polymorphisms and molecular mixtures present in the sample, and putative insertions can be observed more clearly. An ideal high accuracy basecall will have a sharp, distinct peak of a single color indicating a single nucleotide, whereas a low accuracy basecall will have a less defined or mixed (overlapping) peaks.
For plasmids, "failure" means that your sample did not produce data of sufficient quality and quantity for the pipeline to generate a consensus sequence.
Our low sequencing prices and fast turnaround times do not include extensive QC to determine why your plasmid samples failed (or had low coverage). Although we do not provide definitive reasons for failure, by far the most common reasons are:
- Samples are not prepared at the required DNA concentration.
The most common cause of this is using a Nanodrop to quantify DNA concentration. We strongly recommend using a Qubit or equivalent.
You may see evidence of this failure mode in the low amount of total data reported in the raw read length histogram and in the low consensus coverage reported in the SAMPLE_summary.tsv file. - Samples contain a mixture of plasmid species and/or fragmented genomic DNA or fragmented plasmids.
You may see evidence of this failure mode in a wide range of read lengths reported in the raw read length histogram.
To achieve optimal sequencing results, please follow our recommended plasmid sample prep instructions, ZeroPrep cell prep instructions, or RCA sample prep instructions
It is relatively rare that we cannot return a consensus sequence, but some rate of failure is unavoidable. You are welcome to submit a rerun request for any failed plasmid samples through your Order Info page (please note that ZeroPrep and RCA Sequencing Services are NOT eligible for reruns). We will evaluate whether your plasmid sample quality and quantity permits rerunning your sample (we may also ask you to provide a reference sequence). We do still charge for failed samples.
When you upload a reference sequence, we use Minimap to determine the alignment of your assembly to the reference sequence you provided.
If your sample is a “likely match”, this means that all the mismatches in your sample fall into a pattern common to ONT sequencing artifacts. These are DNA methylation sites, and insertions and deletions in long runs of the same nucleotide (homopolymers, e.g. A10->A9).
If your sample is a “mismatch”, it contains mismatches that do not fall into ONT sequencing artifact patterns.
If your sample is “no match”, this means we did not find any alignment between your assembly and your uploaded references.